Phage display of macrocyclic peptide libraries has become a reliable route to high-affinity ligands for difficult protein targets, yet the field has largely treated target immobilization as a technical footnote rather than a scientific variable. Conventional strategies, including immunocapture, biotin–streptavidin capture, and NHS-activated direct coupling, each carry a structural cost: random labeling on lysine residues can disrupt salt bridges, occlude binding epitopes, or lock a protein in a non-native orientation. After more than three decades of biopanning campaigns, no systematic study had quantified how the structural integrity of an immobilized target shapes the identity, affinity, and enrichment efficiency of discovered macrocyclic peptide binders.
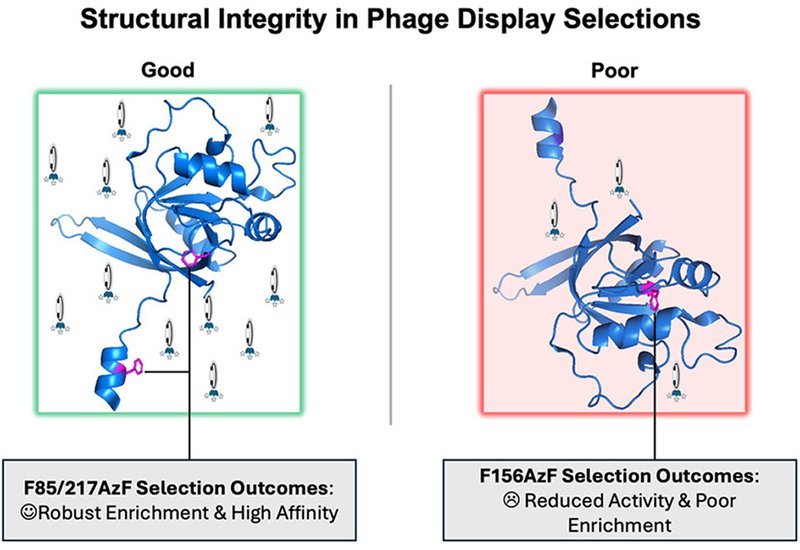
Researchers in the Liu and Hampton Groups at Texas A&M University, published in ACS Chemical Biology, used the ZNRF3 ectodomain as a model to fill that gap. The team employed genetic code expansion to replace three phenylalanine residues, F217, F85, and F156, with p-azidophenylalanine, AzF, chosen because each site carries a distinct solvent exposure: F217 sits on a flexible, fully exposed loop; F85 is partially buried with its side chain engaged in local contacts; and F156 lies deep within the folded β-sheet core. Each AzF mutant was site-specifically conjugated to dibenzocyclooctyne-functionalized magnetic beads via copper-free strain-promoted azide–alkyne cycloaddition, SPAAC, giving three versions of the same target anchored with progressively greater structural disruption. A CX12C disulfide-cyclized phage library was then panned against each variant across three rounds of selection and analyzed by next-generation sequencing.
The structural consequences tracked closely with burial depth. Biolayer interferometry showed that F217AzF retained near-wild-type affinity for R-spondin 2, with a KD of 42 nM against 47 nM for the wild-type protein, while F85AzF and F156AzF displayed roughly 7-fold and 9-fold reduced affinity. Circular dichroism confirmed that all variants retained global secondary structure, though F156AzF showed deviations consistent with perturbation of the β-sheet core. Phage enrichment mirrored these differences: F217AzF produced 45 sequences above 0.1% abundance in round 3, F85AzF produced 27, and F156AzF only 4. The two intact variants shared 2,580 sequences, more than 10% of the sequencing population, while overlap with F156AzF was sparse. NHS-based lysine coupling of wild-type ZNRF3 produced an enrichment profile as diffuse as F156AzF, showing that structure-blind immobilization carries a cost comparable to burying the attachment point in the protein core.
Structural preservation also translated into higher hit rates against native ZNRF3. Among the top five sequences from each variant, F217AzF and F85AzF yielded binders at rates of 3/5 and 2/5, while F156AzF produced only 1/5. Peptides drawn from any two-variant overlap proved especially productive, with 9 of 12 such sequences showing measurable affinity. The highest-affinity ligand of the entire study, peptide 85-2, emerged from the all-three common pool at a KD of 124 nM. Notably, the single most abundant sequence across all three selections proved to be a nonspecific binder, underscoring the residual risk of artifact enrichment even when negative selection steps are included.
The study delivers a practical framework for phage display practitioners: site-specific immobilization that preserves the native fold consistently outperforms structure-blind approaches in both enrichment efficiency and the probability of recovering biologically relevant binders. The SPAAC-based approach also offers a tunable probe for mapping which regions of a given target tolerate surface tethering. The authors draw a direct parallel to peptide immunogen strategies in antibody discovery, where structurally decoupled fragments frequently elicit binders that fail to recognize the full-length antigen, and caution that fragment-based phage selections carry an analogous structural fidelity cost.